A Quelques formats de données en biologie¶
A.1 FASTA¶
Le format FASTA est utilisé pour stocker une ou plusieurs séquences, d'ADN, d'ARN ou de protéines. Ces séquences sont classiquement représentées sous la forme :
>en-tête
séquence avec un nombre maximum de caractères par ligne
séquence avec un nombre maximum de caractères par ligne
séquence avec un nombre maximum de caractères par ligne
séquence avec un nombre maximum de caractères par ligne
séquence avec un nombre max
La première ligne débute par le caractère > et contient une description de la séquence. On appelle souvent cette ligne « ligne de description » ou « ligne de commentaire ».
Les lignes suivantes contiennent la séquence à proprement dite, mais avec un nombre maximum fixe de caractères par ligne. Ce nombre maximum est généralement fixé à 60, 70 ou 80 caractères. Une séquence de plusieurs centaines de bases ou de résidus est donc répartie sur plusieurs lignes.
Un fichier est dit multifasta lorsqu'il contient plusieurs séquences au format FASTA, les unes à la suite des autres.
Les fichiers contenant une ou plusieurs séquences au format FASTA portent la plupart du temps l'extension .fasta mais on trouve également .seq, .fas, .fna ou .faa.
A.1.1 Exemples¶
La séquence protéique au format FASTA de l'insuline humaine, extraite de la base de données UniProt, est :
>sp|P01308|INS_HUMAN Insulin OS=Homo sapiens OX=9606 GN=INS PE=1 SV=1
MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAED
LQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
La première ligne contient la description de la séquence (Insulina), le type de base de données (ici, sp qui signifie Swiss-Prot), son identifiant (P01308) et son nom (INS_HUMAN) dans cette base de données, ainsi que d'autres informations (OS=Homo sapiens OX=9606 GN=INS PE=1 SV=1B).
Les lignes suivantes contiennent la séquence sur des lignes ne dépassant pas, ici, 60 caractères. La séquence de l'insuline humaine est composée de 110 acides aminés, soit une ligne de 60 caractères et une seconde de 50 caractères.
UniProt est une base de données de séquences de protéines. Ces séquences proviennent elles-mêmes de deux autres bases de données : Swiss-Prot (où les séquences sont annotées manuellement) et TrEMBL (où les séquences sont annotées automatiquement).
Voici maintenant la séquence nucléique (ARN), au format FASTA, de l'insuline humaine, extraite de la base de données GenBank :
>BT006808.1 Homo sapiens insulin mRNA, complete cds
ATGGCCCTGTGGATGCGCCTCCTGCCCCTGCTGGCGCTGCTGGCCCTCTGGGGACCTGACCCAGCCGCAG
CCTTTGTGAACCAACACCTGTGCGGCTCACACCTGGTGGAAGCTCTCTACCTAGTGTGCGGGGAACGAGG
CTTCTTCTACACACCCAAGACCCGCCGGGAGGCAGAGGACCTGCAGGTGGGGCAGGTGGAGCTGGGCGGG
GGCCCTGGTGCAGGCAGCCTGCAGCCCTTGGCCCTGGAGGGGTCCCTGCAGAAGCGTGGCATTGTGGAAC
AATGCTGTACCAGCATCTGCTCCCTCTACCAGCTGGAGAACTACTGCAACTAG
On retrouve sur la première ligne la description de la séquence (Homo sapiens insulin mRNA), ainsi que son identifiant (BT006808.1) dans la base de données GenBank.
Les lignes suivantes contiennent les 333 bases de la séquence, réparties sur cinq lignes de 70 caractères maximum. Il est curieux de trouver la base T (thymine) dans une séquence d'ARN, qui ne devrait contenir normalement que les bases A, U, G et C. Ici, la représentation d'une séquence d'ARN avec les bases de l'ADN est une convention.
Pour terminer, voici trois séquences protéiques, au format FASTA, qui correspondent à l'insuline humaine (Homo sapiens), féline (Felis catus) et bovine (Bos taurus) :
>sp|P01308|INS_HUMAN Insulin OS=Homo sapiens OX=9606 GN=INS PE=1 SV=1
MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAED
LQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
>sp|P06306|INS_FELCA Insulin OS=Felis catus OX=9685 GN=INS PE=1 SV=2
MAPWTRLLPLLALLSLWIPAPTRAFVNQHLCGSHLVEALYLVCGERGFFYTPKARREAED
LQGKDAELGEAPGAGGLQPSALEAPLQKRGIVEQCCASVCSLYQLEHYCN
>sp|P01317|INS_BOVIN Insulin OS=Bos taurus OX=9913 GN=INS PE=1 SV=2
MALWTRLRPLLALLALWPPPPARAFVNQHLCGSHLVEALYLVCGERGFFYTPKARREVEG
PQVGALELAGGPGAGGLEGPPQKRGIVEQCCASVCSLYQLENYCN
Ces séquences proviennent de la base de données UniProt.
Chaque séquence est délimitée par la ligne d'en-tête qui débute par >.
A.1.2 Manipulation avec Python¶
À partir de l'exemple précédent des 3 séquences d'insuline, voici un exemple de code qui lit un fichier FASTA avec Python :
prot_dict = {}
with open("insulin.fasta", "r") as fasta_file:
prot_id = ""
for line in fasta_file:
if line.startswith(">"):
prot_id = line[1:].split()[0]
prot_dict[prot_id] = ""
else:
prot_dict[prot_id] += line.strip()
for id in prot_dict:
print(id)
print(prot_dict[id][:30])
Pour chaque séquence lue dans le fichier FASTA, on affiche son identifiant et son nom, puis les 30 premiers résidus de sa séquence :
sp|P06306|INS_FELCA
MAPWTRLLPLLALLSLWIPAPTRAFVNQHL
sp|P01317|INS_BOVIN
MALWTRLRPLLALLALWPPPPARAFVNQHL
sp|P01308|INS_HUMAN
MALWMRLLPLLALLALWGPDPAAAFVNQHL
Notez que les protéines sont stockées dans un dictionnaire (prot_dict) où les clefs sont les identifiants et les valeurs les séquences.
On peut faire la même chose avec le module Biopython :
from Bio import SeqIO
with open("insulin.fasta", "r") as fasta_file:
for record in SeqIO.parse(fasta_file, "fasta"):
print(record.id)
print(str(record.seq)[:30])
Cela produit le même résultat. L'utilisation de Biopython rend le code plus compacte car on utilise ici la fonction SeqIO.parse() qui s'occupe de lire le fichier FASTA.
L'attribut .id renvoie l'identifiant d'une séquence, c'est-à-dire la première partie de l'entête, sans le caractère >. Pour obtenir l'entête complet (toujours sans le caractère >), il faut utiliser l'attribut .description.
A.2 GenBank¶
GenBank est une banque de séquences nucléiques. Le format de fichier associé contient l'information nécessaire pour décrire un gène ou une portion d'un génome. Les fichiers GenBank portent le plus souvent l'extension .gbk.
Le format GenBank est décrit de manière très complète sur le site du NCBI. En voici néanmoins les principaux éléments, avec l'exemple du gène qui code pour la trypsine chez l'Homme.
A.2.1 L'en-tête¶
LOCUS HUMTRPSGNA 800 bp mRNA linear PRI 14-JAN-1995
DEFINITION Human pancreatic trypsin 1 (TRY1) mRNA, complete cds.
ACCESSION M22612
VERSION M22612.1
KEYWORDS trypsinogen.
SOURCE Homo sapiens (human)
ORGANISM Homo sapiens
Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi;
Mammalia; Eutheria; Euarchontoglires; Primates; Haplorrhini;
Catarrhini; Hominidae; Homo.
[...]
- Ligne 1 (
LOCUS) : le nom du locus (HUMTRPSGNA), la taille du gène (800 paires de bases), le type de molécule (ARN messager). - Ligne 3 (
ACCESSION) : l'identifiant de la séquence (M22612). - Ligne 4 (
VERSION) : la version de la séquence (M22612.1). Le nombre qui est séparé de l'identifiant de la séquence par un point est incrémenté pour chaque nouvelle version de la fiche GenBank. Ici, .1 indique que nous en sommes à la première version. - Ligne 6 (
SOURCE) : la provenance de la séquence (souvent l'organisme d'origine). - Ligne 7 (
ORGANISME) : le nom scientifique de l'organisme, suivi de sa taxonomie (lignes 8 à 10).
A.2.2 Les features¶
[...]
FEATURES Location/Qualifiers
source 1..800
/organism="Homo sapiens"
/mol_type="mRNA"
/db_xref="taxon:9606"
/map="7q32-qter"
/tissue_type="pancreas"
gene 1..800
/gene="TRY1"
CDS 7..750
/gene="TRY1"
/codon_start=1
/product="trypsinogen"
/protein_id="AAA61231.1"
/db_xref="GDB:G00-119-620"
/translation="MNPLLILTFVAAALAAPFDDDDKIVGGYNCEENSVPYQVSLNSG
YHFCGGSLINEQWVVSAGHCYKSRIQVRLGEHNIEVLEGNEQFINAAKIIRHPQYDRK
TLNNDIMLIKLSSRAVINARVSTISLPTAPPATGTKCLISGWGNTASSGADYPDELQC
LDAPVLSQAKCEASYPGKITSNMFCVGFLEGGKDSCQGDSGGPVVCNGQLQGVVSWGD
GCAQKNKPGVYTKVYNYVKWIKNTIAANS"
sig_peptide 7..51
/gene="TRY1"
/note="G00-119-620"
[...]
- Ligne 9 (
gene 1..800) : la délimitation du gène. Ici, de la base 1 à la base 800. Par ailleurs, la notation<x..yindique que la séquence est partielle sur l'extrémité 5'. Réciproquement,x..y>indique que la séquence est partielle sur l'extrémité 3'. Enfin, pour les séquences d'ADN, la notationcomplement(x..y)indique que le gène se trouve de la base x à la base y, mais sur le brin complémentaire. - Ligne 10 (
/gene="TRY1") : le nom du gène. - Ligne 11 (
CDS 7..750) : la délimitation de la séquence codante. - Ligne 14 (
/product="trypsinogen") : le nom de la protéine produite. - Lignes 17 à 20 (
/translation="MNPLLIL...) : la séquence protéique issue de la traduction de la séquence codante. - Ligne 22 (
sig_peptide 7..51) : la délimitation du peptide signal.
A.2.3 La séquence¶
[...]
ORIGIN
1 accaccatga atccactcct gatccttacc tttgtggcag ctgctcttgc tgcccccttt
61 gatgatgatg acaagatcgt tgggggctac aactgtgagg agaattctgt cccctaccag
121 gtgtccctga attctggcta ccacttctgt ggtggctccc tcatcaacga acagtgggtg
181 gtatcagcag gccactgcta caagtcccgc atccaggtga gactgggaga gcacaacatc
241 gaagtcctgg aggggaatga gcagttcatc aatgcagcca agatcatccg ccacccccaa
301 tacgacagga agactctgaa caatgacatc atgttaatca agctctcctc acgtgcagta
361 atcaacgccc gcgtgtccac catctctctg cccaccgccc ctccagccac tggcacgaag
421 tgcctcatct ctggctgggg caacactgcg agctctggcg ccgactaccc agacgagctg
481 cagtgcctgg atgctcctgt gctgagccag gctaagtgtg aagcctccta ccctggaaag
541 attaccagca acatgttctg tgtgggcttc cttgagggag gcaaggattc atgtcagggt
601 gattctggtg gccctgtggt ctgcaatgga cagctccaag gagttgtctc ctggggtgat
661 ggctgtgccc agaagaacaa gcctggagtc tacaccaagg tctacaacta cgtgaaatgg
721 attaagaaca ccatagctgc caatagctaa agcccccagt atctcttcag tctctatacc
781 aataaagtga ccctgttctc
//
La séquence est contenue entre les balises ORIGIN (ligne 2) et // (ligne 17).
Chaque ligne est composée d'une série d'espaces, puis du numéro du premier nucléotide de la ligne, puis d'au plus 6 blocs de 10 nucléotides. Chaque bloc est précédé d'un espace. Par exemple, ligne 10, le premier nucléotide de la ligne (t) est le numéro 421 dans la séquence.
A.2.4 Manipulation avec Python¶
À partir de l'exemple précédent, voici comment lire un fichier GenBank avec Python et le module Biopython :
from Bio import SeqIO
with open("M22612.gbk", "r") as gbk_file:
record = SeqIO.read(gbk_file, "genbank")
print(record.id)
print(record.description)
print(record.seq[:60])
M22612.1
Human pancreatic trypsin 1 (TRY1) mRNA, complete cds.
ACCACCATGAATCCACTCCTGATCCTTACCTTTGTGGCAGCTGCTCTTGCTGCCCCCTTT
Il est également possible de lire un fichier GenBank sans le module Biopython. Une activité dédiée est proposée dans le chapitre 27 Mini-projets (en ligne).
A.3 PDB¶
La Protein Data Bank (PDB) est une banque de données qui contient les structures de biomacromolécules (protéines, ADN, ARN, virus...). Historiquement, le format de fichier qui y est associé est le PDB, dont une documentation détaillée est disponible sur le site éponyme. Les extensions de fichier pour ce format de données sont .ent et surtout .pdb.
Un fichier PDB est constitué de deux parties principales : l'en-tête et les coordonnées.
- L'en-tête est lisible et utilisable par un être humain (comme par une machine).
- À l'inverse, les coordonnées sont surtout utilisables par un programme pour calculer certaines propriétés de la structure ou simplement la représenter sur l'écran d'un ordinateur. Bien sûr, un utilisateur expérimenté peut parfaitement jeter un œil à cette seconde partie.
Examinons ces deux parties avec la trypsine bovine.
A.3.1 En-tête¶
Pour la trypsine bovine, l'en-tête compte 510 lignes. En voici quelques unes :
HEADER HYDROLASE (SERINE PROTEINASE) 26-OCT-81 2PTN
TITLE ON THE DISORDERED ACTIVATION DOMAIN IN TRYPSINOGEN.
TITLE 2 CHEMICAL LABELLING AND LOW-TEMPERATURE CRYSTALLOGRAPHY
COMPND MOL_ID: 1;
COMPND 2 MOLECULE: TRYPSIN;
COMPND 3 CHAIN: A;
[...]
SOURCE 2 ORGANISM_SCIENTIFIC: BOS TAURUS;
[...]
EXPDTA X-RAY DIFFRACTION
[...]
REMARK 2 RESOLUTION. 1.55 ANGSTROMS.
[...]
DBREF 2PTN A 16 245 UNP P00760 TRY1_BOVIN 21 243
SEQRES 1 A 223 ILE VAL GLY GLY TYR THR CYS GLY ALA ASN THR VAL PRO
SEQRES 2 A 223 TYR GLN VAL SER LEU ASN SER GLY TYR HIS PHE CYS GLY
SEQRES 3 A 223 GLY SER LEU ILE ASN SER GLN TRP VAL VAL SER ALA ALA
SEQRES 4 A 223 HIS CYS TYR LYS SER GLY ILE GLN VAL ARG LEU GLY GLU
[...]
HELIX 1 H1 SER A 164 ILE A 176 1SNGL ALPHA TURN,REST IRREG. 13
HELIX 2 H2 LYS A 230 VAL A 235 5CONTIGUOUS WITH H3 6
HELIX 3 H3 SER A 236 ASN A 245 1CONTIGUOUS WITH H2 10
SHEET 1 A 7 TYR A 20 THR A 21 0
SHEET 2 A 7 LYS A 156 PRO A 161 -1 N CYS A 157 O TYR A 20
[...]
SSBOND 1 CYS A 22 CYS A 157 1555 1555 2.04
SSBOND 2 CYS A 42 CYS A 58 1555 1555 2.02
[...]
- Ligne 1. Cette ligne
HEADERcontient :- le nom de la protéine : HYDROLASE (SERINE PROTEINASE),
- la date de dépôt de cette structure dans la banque de données : 26 octobre 1981
- et l'identifiant de la structure dans la PDB, on parle souvent de « code PDB » : 2PTN.
- Ligne 2.
TITLEcorrespond au titre de l'article scientifique dans lequel a été publié cette structure. - Lignes 4-6.
COMPNDindique que la trypsine est composée d'une seule chaîne peptidique, appelée iciA. - Ligne 8.
SOURCEindique le nom scientifique de l'organisme dont provient cette protéine (ici, le bœuf). - Ligne 10.
EXPDTAprécise la technique expérimentale employée pour déterminer cette structure. Ici, la cristallographie aux rayons X. Mais on peut également trouver SOLUTION NMR pour de la résonance magnétique nucléaire en solution, ELECTRON MICROSCOPY pour de la microscopie électronique. - Ligne 12.
REMARK 2précise, dans le cas d'une détermination par cristallographie aux rayons X, la résolution obtenue, ici 1,55 Angströms. - Ligne 14.
DBREFindique les liens éventuels vers d'autres banques de données. Ici, l'identifiant correspondant à cette protéine dans UniProt (UNP) est P00760. - Ligne 15-18.
SEQRESdonnent à la séquence de la protéine. Les résidus sont représentés par leur code à trois lettres. - Lignes 20-22 et 23-24.
HELIXetSHEETcorrespondent aux structures secondaires hélices \(\alpha\) et brin \(\beta\) de cette protéine. Ici, H1 SER A 164 ILE A 176 indique qu'il y a une première hélice \(\alpha\) (H1), comprise entre les résidus Ser164 et Ile176 de la chaîne A. - Lignes 26-27.
SSBONDindique les ponts disulfures. Ici, entre les résidus Cys22 et Cys157 et entre les résidus Cys42 et Cys58.
A.3.2 Coordonnées¶
Avec la même protéine, la partie coordonnées représente plus de 1 700 lignes. En voici quelques unes correspondantes au résidu leucine 99 :
[...]
ATOM 601 N LEU A 99 10.007 19.687 17.536 1.00 12.25 N
ATOM 602 CA LEU A 99 9.599 18.429 18.188 1.00 12.25 C
ATOM 603 C LEU A 99 10.565 17.281 17.914 1.00 12.25 C
ATOM 604 O LEU A 99 10.256 16.101 18.215 1.00 12.25 O
ATOM 605 CB LEU A 99 8.149 18.040 17.853 1.00 12.25 C
ATOM 606 CG LEU A 99 7.125 19.029 18.438 1.00 18.18 C
ATOM 607 CD1 LEU A 99 5.695 18.554 18.168 1.00 18.18 C
ATOM 608 CD2 LEU A 99 7.323 19.236 19.952 1.00 18.18 C
[...]
Chaque ligne correspond à un atome et débute par ATOM ou HETATM.
ATOM désigne un atome de la structure de la biomolécule.
HETATM est utilisé pour les atomes qui ne sont pas une biomolécule, comme les ions ou les molécules d'eau.
Toutes les lignes de coordonnées ont sensiblement le même format. Par exemple, pour la première ligne :
ATOM(ouHETATM).601: le numéro de l'atome.N: le nom de l'atome. Ici, un atome d'azote du squelette peptidique. La structure complète du résidu leucine est représentée figure 1.LEU: le résidu dont fait partie l'atome. Ici, une leucine.A: le nom de la chaîne peptidique.99: le numéro du résidu dans la protéine.10.007: la coordonnée x de l'atome.19.687: la coordonnée y de l'atome.17.536: la coordonnée z de l'atome.1.00: le facteur d'occupation, c'est-à-dire la probabilité de trouver l'atome à cette position dans l'espace en moyenne. Cette probabilité est inférieure à 1 lorsque, expérimentalement, on n'a pas pu déterminer avec une totale certitude la position de l'atome. Par exemple, dans le cas d'un atome très mobile dans une structure, qui est déterminé comme étant à deux positions possibles, chaque position aura alors la probabilité0.50.12.25: le facteur de température, qui est proportionnel à la mobilité de l'atome dans l'espace. Les atomes situés en périphérie d'une structure sont souvent plus mobiles que ceux situés au coeur de la structure.N: l'élément chimique de l'atome. Ici, l'azote.
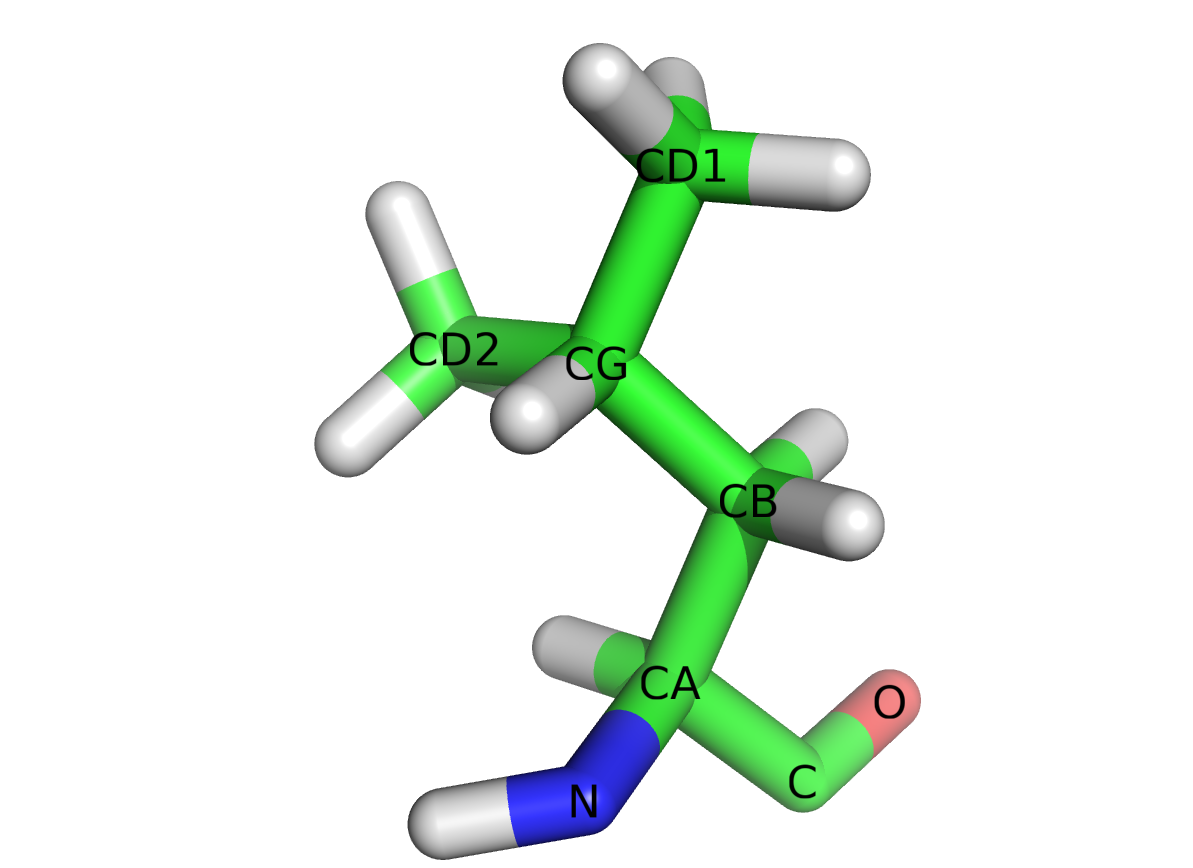
Une documentation plus complète des différents champs qui constituent une ligne de coordonnées atomiques se trouve sur le site de la PDB.
Les résidus sont ensuite décrits les uns après les autres, atome par atome. Voici par exemple les premiers résidus de la trypsine bovine :
[...]
ATOM 1 N ILE A 16 -8.155 9.648 20.365 1.00 10.68 N
ATOM 2 CA ILE A 16 -8.150 8.766 19.179 1.00 10.68 C
ATOM 3 C ILE A 16 -9.405 9.018 18.348 1.00 10.68 C
ATOM 4 O ILE A 16 -10.533 8.888 18.870 1.00 10.68 O
ATOM 5 CB ILE A 16 -8.091 7.261 19.602 1.00 10.68 C
ATOM 6 CG1 ILE A 16 -6.898 6.882 20.508 1.00 7.42 C
ATOM 7 CG2 ILE A 16 -8.178 6.281 18.408 1.00 7.42 C
ATOM 8 CD1 ILE A 16 -5.555 6.893 19.773 1.00 7.42 C
ATOM 9 N VAL A 17 -9.224 9.305 17.090 1.00 9.63 N
ATOM 10 CA VAL A 17 -10.351 9.448 16.157 1.00 9.63 C
ATOM 11 C VAL A 17 -10.500 8.184 15.315 1.00 9.63 C
ATOM 12 O VAL A 17 -9.496 7.688 14.748 1.00 9.63 O
ATOM 13 CB VAL A 17 -10.123 10.665 15.222 1.00 9.63 C
ATOM 14 CG1 VAL A 17 -11.319 10.915 14.278 1.00 11.95 C
ATOM 15 CG2 VAL A 17 -9.737 11.970 15.970 1.00 11.95 C
[...]
Vous remarquerez que le numéro du premier résidu est 16 et non pas 1. Cela s'explique par la technique expérimentale utilisée qui n'a pas permis de déterminer la structure des 15 premiers résidus.
La structure de la trypsine bovine n'est constituée que d'une seule chaîne peptidique (notée A).
Lorsqu'une structure est composée de plusieurs chaînes, comme dans le cas de la structure du récepteur GABAB 1 et 2 chez la drosophile (code PDB 5X9X) :
[...]
ATOM 762 HB1 ALA A 44 37.162 -2.955 2.220 1.00 0.00 H
ATOM 763 HB2 ALA A 44 38.306 -2.353 3.417 1.00 0.00 H
ATOM 764 HB3 ALA A 44 38.243 -1.621 1.814 1.00 0.00 H
TER 765 ALA A 44
ATOM 766 N GLY B 95 -18.564 3.009 13.772 1.00 0.00 N
ATOM 767 CA GLY B 95 -19.166 3.646 12.621 1.00 0.00 C
ATOM 768 C GLY B 95 -20.207 2.755 11.976 1.00 0.00 C
[...]
La première chaîne est notée A et la seconde B. La séparation entre
les deux chaînes est marquée par la ligne :
Dans un fichier PDB, chaque structure porte un nom de chaîne différent (par exemple : A,B,C`, etc.).
Enfin, lorsque la structure est déterminée par RMN, il est possible que plusieurs structures soient présentes dans le même fichier PDB. Toutes ces structures, ou « modèles », sont des solutions possibles du jeu de contraintes mesurées expérimentalement en RMN. Voici un exemple, toujours pour la structure du récepteur GABAB 1 et 2 chez la drosophile :
[...]
MODEL 1
ATOM 1 N MET A 1 -27.283 -9.772 5.388 1.00 0.00 N
ATOM 2 CA MET A 1 -28.233 -8.680 5.682 1.00 0.00 C
[...]
ATOM 1499 HG2 GLU B 139 36.113 -5.242 2.536 1.00 0.00 H
ATOM 1500 HG3 GLU B 139 37.475 -4.132 2.428 1.00 0.00 H
TER 1501 GLU B 139
ENDMDL
MODEL 2
ATOM 1 N MET A 1 -29.736 -10.759 4.394 1.00 0.00 N
ATOM 2 CA MET A 1 -28.372 -10.225 4.603 1.00 0.00 C
[...]
ATOM 1499 HG2 GLU B 139 36.113 -5.242 2.536 1.00 0.00 H
ATOM 1500 HG3 GLU B 139 37.475 -4.132 2.428 1.00 0.00 H
TER 1501 GLU B 139
ENDMDL
MODEL 2
ATOM 1 N MET A 1 -29.736 -10.759 4.394 1.00 0.00 N
ATOM 2 CA MET A 1 -28.372 -10.225 4.603 1.00 0.00 C
[...]
Chaque structure est encadrée par les lignes :
et :
où n est le numéro du modèle. Pour la structure du récepteur GABAB 1 et 2, il y a 20 modèles de décrits dans le fichier PDB.
A.3.3 Manipulation avec Python¶
Le module Biopython peut également lire un fichier PDB.
Voici comment charger la structure de la trypsine bovine :
from Bio.PDB import PDBParser
parser = PDBParser()
prot_id = "2PTN"
prot_file = "2PTN.pdb"
structure = parser.get_structure(prot_id, prot_file)
Les fichiers PDB sont parfois (très) mal formatés. Si Biopython ne parvient pas à lire un tel fichier,
remplacez alors la ligne 2 par parser = PDBParser(PERMISSIVE=1). Soyez néanmoins très prudent quant aux résultats obtenus.
Affichage du nom de la structure et de la technique expérimentale utilisée pour déterminer la structure :
ce qui produit :
Extraction des coordonnées de l'atome N du résidu Ile16 et de l'atome CA du résidu Val17 :
model = structure[0]
chain = model["A"]
res1 = chain[16]
res2 = chain[17]
print(res1.resname, res1["N"].coord)
print(res2.resname, res2["CA"].coord)
ce qui produit :
L'objet res1["N"].coord est un array de NumPy (voir le chapitre 20
Module NumPy). On peut alors obtenir
simplement les coordonnées x, y et z d'un atome :
ce qui produit :
Biopython utilise la hiérarchie suivante :
structure > model > chain > residue > atom
même lorsque la structure ne contient qu'un seul modèle. C'est d'ailleurs
le cas ici, puisque la structure
a été obtenue par cristallographie aux rayons X.
Enfin, pour afficher les coordonnées des carbones \(\alpha\) (notés CA) des 10 premiers résidus (à partir du résidu 16, car c'est le premier résidu dont on connaît la structure) :
res_start = 16
model = structure[0]
chain = model["A"]
for i in range(10):
idx = res_start + i
print(chain[idx].resname, idx, chain[idx]["CA"].coord)
avec pour résultat :
ILE 16 [ -8.14999962 8.76599979 19.17900085]
VAL 17 [-10.35099983 9.44799995 16.15699959]
GLY 18 [-12.02099991 6.63000011 14.25899982]
GLY 19 [-10.90200043 3.89899993 16.68400002]
TYR 20 [-12.65100002 1.44200003 19.01600075]
THR 21 [-13.01799965 0.93800002 22.76000023]
CYS 22 [-10.02000046 -1.16299999 23.76000023]
GLY 23 [-11.68299961 -2.86500001 26.7140007 ]
ALA 24 [-10.64799976 -2.62700009 30.36100006]
ASN 25 [ -6.96999979 -3.43700004 31.02000046]
Il est aussi très intéressant (et formateur) d'écrire son propre parser de fichier PDB,
c'est-à-dire un programme qui lit un fichier PDB (sans le module Biopython).
Dans ce cas, la figure 2 vous aidera à déterminer comment extraire
les différentes informations d'une ligne de coordonnées ATOM ou HETATM.
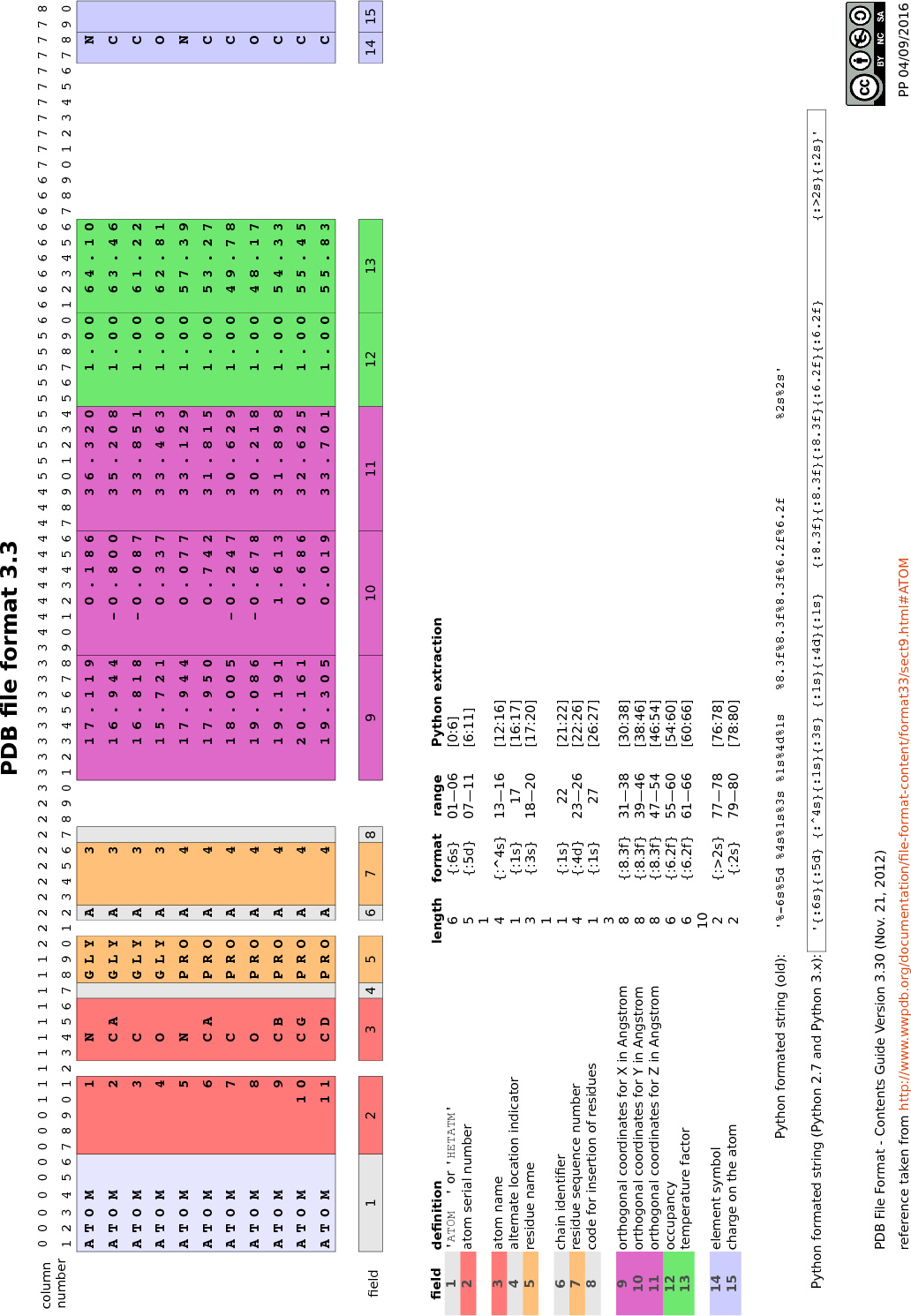
Exemple : pour extraire le nom du résidu, il faut isoler le contenu des colonnes 18 à 20 du fichier PDB, ce qui correspond aux index de 17 à 19 pour une chaîne de caractères en Python
(soit la tranche de chaîne de caractères [17:20], car la première borne est incluse et la seconde exclue).
Pour lire le fichier PDB de la trypsine bovine (2PTN.pdb) et extraire (encore) les coordonnées des carbones \(\alpha\) des 10 premiers résidus, nous pouvons utiliser le code suivant :
with open("2PTN.pdb", "r") as pdb_file:
res_count = 0
for line in pdb_file:
if line.startswith("ATOM"):
atom_name = line[12:16].strip()
res_name = line[17:20].strip()
res_num = int(line[22:26])
if atom_name == "CA":
res_count += 1
x = float(line[30:38])
y = float(line[38:46])
z = float(line[46:54])
print(res_name, res_num, x, y, z)
if res_count >= 10:
break
ce qui donne :
ILE 16 -8.15 8.766 19.179
VAL 17 -10.351 9.448 16.157
GLY 18 -12.021 6.63 14.259
GLY 19 -10.902 3.899 16.684
TYR 20 -12.651 1.442 19.016
THR 21 -13.018 0.938 22.76
CYS 22 -10.02 -1.163 23.76
GLY 23 -11.683 -2.865 26.714
ALA 24 -10.648 -2.627 30.361
ASN 25 -6.97 -3.437 31.02
Pour extraire des valeurs numériques, comme des numéros de résidus ou des coordonnées atomiques, il ne faudra pas oublier de les convertir en entiers ou en floats.
A.4 Format XML, CSV et TSV¶
Les formats XML, CSV et TSV dont des formats de fichiers très largement utilisés en informatique. Ils sont également très utilisés en biologie. En voici quelques exemples :
A.4.1 XML¶
Le format XML est un format de fichier à balises qui permet de stocker quasiment n'importe quel type d'information de façon structurée et hiérarchisée. L'acronyme XML signifie Extensible Markup Language qui pourrait se traduire en français par « Langage de balisage extensible ». Les balises dont il est question servent à délimiter du contenu :
La balise <balise> est une balise ouvrante.
La balise </balise> est une balise fermante.
Notez le caractère / qui marque la différence entre une balise ouvrante
et une balise fermante.
Il existe également des balises vides, qui sont à la fois ouvrantes et fermantes :
Une balise peut avoir certaines propriétés, appelées attributs, qui sont définies, dans la balise ouvrante. Par exemple :
Un attribut est un couple nom et valeur (par exemple propriété1 est un nom
et valeur1 est la valeur associée).
Enfin, les balises peuvent être imbriquées les unes dans les autres :
<protein>
<element>élément 1</element>
<element>élément 2</element>
<element>élément 3</element>
</protein>
Dans cet exemple, nous avons trois balises element qui sont contenues dans une balise
protein.
Voici un autre exemple avec l'enzyme trypsine humaine (code P07477), telle qu'on peut la trouver décrite dans la base de données UniProt :
<?xml version='1.0' encoding='UTF-8'?>
<uniprot xmlns="http://uniprot.org/uniprot" xmlns:xsi=[...]>
<entry dataset="Swiss-Prot" created="1988-04-01" modified="2018-09-12" [...]>
<accession>P07477</accession>
<accession>A1A509</accession>
[...]
<gene>
<name type="primary">PRSS1</name>
<name type="synonym">TRP1</name>
<name type="synonym">TRY1</name>
</gene>
[...]
<sequence length="247" mass="26558" checksum="DD49A487B8062813" [...]>
MNPLLILTFVAAALAAPFDDDDKIVGGYNCEENSVPYQVSLNSGYHFCGGSLINEQWVVS
AGHCYKSRIQVRLGEHNIEVLEGNEQFINAAKIIRHPQYDRKTLNNDIMLIKLSSRAVIN
ARVSTISLPTAPPATGTKCLISGWGNTASSGADYPDELQCLDAPVLSQAKCEASYPGKIT
SNMFCVGFLEGGKDSCQGDSGGPVVCNGQLQGVVSWGDGCAQKNKPGVYTKVYNYVKWIK
NTIAANS
</sequence>
</entry>
[...]
</uniprot>
- La ligne 1 indique que nous avons bien un fichier au format XML.
- La ligne 3 indique que nous avons une entrée UniProt. Il s'afit d'une balise
ouvrante avec plusieurs attributs (
dataset="Swiss-Prot",created="1988-04-01", etc.). - Les lignes 4 et 5 précisent les numéros d'accession dans la base de données UniProt qui font référence à cette même protéine.
- Les lignes 8-10 listent les quatre gènes correspondants à cette protéine.
Le premier gène porte l'attribut
type="primary"et indique qu'il s'agit du nom officiel du gène de la trypsine. L'attributtype="synonym"pour les autres gènes indique qu'il s'agit bien de noms synonymes pour le gènePRSS1. - Les lignes 13-18 contiennent la séquence de la trypsine. Dans les attributs de
la balise
<sequence>, on retrouve, par exemple, la taille de la protéine (length="247").
Voici un exemple de code Python pour manipuler le fichier XML de la trypsine humaine :
from lxml import etree
import re
with open("P07477.xml") as xml_file:
xml_content = xml_file.read()
xml_content = re.sub("<uniprot [^>]+>", "<uniprot>", xml_content)
root = etree.fromstring(xml_content.encode("utf-8"))
for gene in root.xpath("/uniprot/entry/gene/name"):
print(f"gene : {gene.text} ({gene.get('type')})")
sequence = root.xpath("/uniprot/entry/sequence")[0]
print(f"sequence: {sequence.text.strip()}")
print(f"length: {sequence.get('length')}")
- Ligne 1. On utilise le sous-module
etreedu module lxml pour lire le fichier XML. - Ligne 2. On utilise le module d'expressions régulières re pour supprimer
tous les attributs de la balise
uniprot(ligne 7). Nous ne rentrerons pas dans les détails, mais ces attributs rendent plus complexe la lecture du fichier XML. - Ligne 9. La variable
rootcontient le fichier XML prêt à être manipulé. - Ligne 11. On recherche les noms des gènes (balises
<name></name>) associés à la trypsine. Pour cela, on utilise la méthode.xpath(), avec comme argument l'enchaînement des différentes balises qui conduisent aux noms des gènes. - Ligne 12. Pour chaque nom de gène, on va afficher son contenu (
gene.text) et la valeur associée à l'attributtypeavec la méthode.get("type"). - Ligne 14. On stocke dans la variable
sequencela balise associée à la séquence de la protéine. Commeroot.xpath("/uniprot/entry/sequence")renvoie un itérateur et qu'il n'y a qu'une seule balise séquence, on prend ici le seul et unique élémentroot.xpath("/uniprot/entry/sequence")[0]. - Ligne 15. On affiche le contenu de la séquence
sequence.text, nettoyé d'éventuels retours chariots ou espacessequence.text.strip(). - Ligne 16. On affiche la taille de la séquence en récupérant la valeur
de l'attribut
length(toujours de la balise<sequence></sequence>).
Le résultat obtenu est le suivant :
gene : PRSS1 (primary)
gene : TRP1 (synonym)
gene : TRY1 (synonym)
gene : TRYP1 (synonym)
sequence: MNPLLILTFVAAALAAPFDDDDKIVGGYNCEENSVPYQVSLNSGYHFCGGSLINEQWVVS
AGHCYKSRIQVRLGEHNIEVLEGNEQFINAAKIIRHPQYDRKTLNNDIMLIKLSSRAVIN
ARVSTISLPTAPPATGTKCLISGWGNTASSGADYPDELQCLDAPVLSQAKCEASYPGKIT
SNMFCVGFLEGGKDSCQGDSGGPVVCNGQLQGVVSWGDGCAQKNKPGVYTKVYNYVKWIK
NTIAANS
length: 247
A.4.2 CSV et TSV¶
Définition des formats¶
L'acronyme CSV signifie « Comma-Separated values », qu'on peut traduire littéralement par « valeurs séparées par des virgules ». De façon similaire, TSV signifie « Tabulation-Separated Values », soit des « valeurs séparées par des tabulations ».
Ces deux formats sont utiles pour stocker des données structurées sous forme de tableau, comme vous pourriez l'avoir dans un tableur.
À titre d'exemple, le tableau ci-dessous liste les structures associées à la transferrine, protéine présente dans le plasma sanguin et impliquée dans la régulation du fer. Ces données proviennent de la Protein Data Bank (PDB). Pour chaque protéine (PDB ID) est indiqué le nom de l'organisme associé (Source), la date à laquelle cette structure a été déposée dans la PDB (Deposit Date), le nombre d'acides aminés de la protéine et sa masse moléculaire (MW).
| PDB ID | Source | Deposit Date | Length | MW |
|---|---|---|---|---|
| 1A8E | Homo sapiens | 1998-03-24 | 329 | 36408.40 |
| 1A8F | Homo sapiens | 1998-03-25 | 329 | 36408.40 |
| 1AIV | Gallus gallus | 1997-04-28 | 686 | 75929.00 |
| 1AOV | Anas platyrhynchos | 1996-12-11 | 686 | 75731.80 |
| [...] | [...] | [...] | [...] | [...] |
Voici maintenant l'équivalent en CSV :
PDB ID,Source,Deposit Date,Length,MW
1A8E,Homo sapiens,1998-03-24,329,36408.40
1A8F,Homo sapiens,1998-03-25,329,36408.40
1AIV,Gallus gallus,1997-04-28,686,75929.00
1AOV,Anas platyrhynchos,1996-12-11,686,75731.80
[...]
Sur chaque ligne, les différentes valeurs sont séparées par une virgule. La première ligne contient le nom des colonnes et est appelée ligne d'en-tête.
L'équivalent en TSV est :
PDB ID Source Deposit Date Length MW
1A8E Homo sapiens 1998-03-24 329 36408.40
1A8F Homo sapiens 1998-03-25 329 36408.40
1AIV Gallus gallus 1997-04-28 686 75929.00
1AOV Anas platyrhynchos 1996-12-11 686 75731.80
[...]
Sur chaque ligne, les différentes valeurs sont séparées par une tabulation.
Le caractère tabulation est un caractère invisible « élastique », c'est-à-dire qu'il a une largeur variable suivant l'éditeur de texte utilisé. Par exemple, dans la ligne d'en-tête, l'espace entre PDB ID et Source apparaît comme différent de l'espace entre Deposit Date et Length alors qu'il y a pourtant une seule tabulation à chaque fois.
Lecture¶
En Python, le module csv de la bibliothèque standard est très pratique pour lire et écrire des fichiers au format CSV et TSV. Nous vous conseillons de lire la documentation très complète sur ce module.
Voici un exemple :
import csv
with open("transferrin_report.csv") as f_in:
f_reader = csv.DictReader(f_in)
for row in f_reader:
print(row["PDB ID"], row["Deposit Date"], row["Length"])
- Ligne 1. Chargement du module csv.
- Ligne 3. Ouverture du fichier.
- Ligne 4. Utilisation du module csv pour lire le fichier CSV comme un dictionnaire (fonction
DictReader()). La ligne d'en-tête est utilisée automatiquement pour définir les clés du dictionnaire. - Ligne 5. Parcours de toutes les lignes du fichier CSV.
- Ligne 6. Affichage des champs correspondants à PDB ID, Deposit Date, Length.
Le résultat obtenu est :
Il suffit de modifier légèrement le script précédent pour lire un fichier TSV :
import csv
with open("transferrin_PDB_report.tsv") as f_in:
f_reader = csv.DictReader(f_in, delimiter="\t")
for row in f_reader:
print(row["PDB ID"], row["Deposit Date"], row["Length"])
- Ligne 3. Modification du nom du fichier lu.
- Ligne 4. Utilisation de l'argument
delimiter="\t", qui indique que les champs sont séparés par des tabulations.
Le résultat obtenu est strictement identique au précédent.
Écriture¶
Voici un exemple d'écriture de fichier CSV :
import csv
with open("test.csv", "w") as f_out:
fields = ["Name", "Quantity"]
f_writer = csv.DictWriter(f_out, fieldnames=fields)
f_writer.writeheader()
f_writer.writerow({"Name": "girafe", "Quantity":5})
f_writer.writerow({"Name": "tigre", "Quantity":3})
f_writer.writerow({"Name": "singe", "Quantity":8})
- Ligne 3. Ouverture du fichier
test.csven lecture. - Ligne 4. Définition du nom des colonnes (Name et Quantity).
- Ligne 5. Utilisation du module csv pour écrire un fichier CSV à partir d'un dictionnaire.
- Ligne 6. Écriture des noms des colonnes.
- Ligne 7-9. Écriture de trois lignes. Pour chaque ligne, un dictionnaire dont les clefs sont les noms des colonnes est fourni comme argument à la méthode
.writerow().
Le contenu du fichier test.csv est alors :
De façon très similaire, l'écriture d'un fichier TSV est réalisée avec le code suivant :
import csv
with open("test.tsv", "w") as f_out:
fields = ["Name", "Quantity"]
f_writer = csv.DictWriter(f_out, fieldnames=fields, delimiter="\t")
f_writer.writeheader()
f_writer.writerow({"Name": "girafe", "Quantity":5})
f_writer.writerow({"Name": "tigre", "Quantity":3})
f_writer.writerow({"Name": "singe", "Quantity":8})
- Ligne 3. Modification du nom du fichier en écriture.
- Ligne 5. Utilisation de l'argument
delimiter="\t", qui indique que les champs sont séparés par des tabulations.
Le contenu du fichier test.tsv est :
Vous êtes désormais capables de lire et écrire des fichiers aux formats CSV et TSV. Les codes que nous vous avons proposés ne sont que des exemples. À vous de poursuivre l'exploration du module csv.
Le module pandas décrit dans le chapitre 22 Module Pandas est tout à fait capable de lire et écrire des fichiers CSV et TSV. Nous vous conseillons de l'utiliser si vous analysez des données avec ces types de fichiers.